

Ask Almost A Doctor: The World Is An LLM Edition
Edition Three


If you have questions, you can email me at eryneym@gmail.com, DM me on Twitter or Substack. Or put them in the comments below!
Also, none of the below constitutes medical advice. (Seriously. This is not medical advice - Ed.)
Oh, and thanks to Kylie Robison for editing.
Enjoy.
Marshm @marshm1 (via Substack)
What is the current state of virtual cells and working all the way up to virtual bodies that serve as useful models to test out new drugs, treatments etc? Can we ever really simulate biology bottom up in enough detail to be useful?
It is not feasible for me to answer whether cell foundation models are ever going to be useful. A better question is to ask what kinds of things these models can be useful for.
Invention shapes how we see the world, so naturally everything has to like, totally be a language model, man. For readers who don’t know, virtual cells and cell foundation models are AI systems trained on single cell (mostly RNA sequencing) data with the aim of learning some latent “language” of biology. These models are built on two types of data: massive atlases of cells just existing in their natural state, and perturbation data. The second kind of data is generated by taking cells, applying some condition (a drug, a CRISPR edit, environmental variable, etc), and sequencing them to see how they react. Don’t call it perturb-seq, though, since that’ll upset cell model makers. It’s important you know that this is a special thing.
I didn’t really have an answer prior to your question, but this was a great excuse to do a little experimentation to answer a very narrow aspect. The short answer, if you want to skip forward through a ton of work I did with Dr. Claude and Dr. Codex, is that I think eventually virtual cells will amount to something of substance for specific questions – like whether some binding event happens – just not every question.
I’m spending the next few months doing various things in clinical oncology, so I was curious to know whether current frontier cell foundation models could answer something pertinent to cancer. There’s a drug called Dabrafenib. It targets a mutation in the BRAF gene, V600E, that shows up in about half of all melanomas and a meaningful chunk of colorectal cancers. In melanoma, it works well enough that it got FDA approval. Response rates around 50%. In colorectal cancer with the exact same mutation, it basically does nothing (response rates around 5%).
Could a frontier virtual cell model have predicted these outcomes before a trial?
The model I used to dig into this is STATE from the Arc Institute, trained on the Tahoe dataset of drug perturbation responses across hundreds of conditions. You give it a cell’s baseline expression profile across 2,000 highly variable genes and a drug label, and it outputs a predicted post-treatment expression profile. I (with help from Drs. Claude Code Max and Codex) pulled the baseline single-cell RNA data from CellxGene Census composed of 300 melanoma cells from skin biopsies and 300 colorectal cancer cells filtered to primary tumors. The measurement is a delta that captures predicted post-drug expression minus a control, log-normalized, averaged across all 2,000 genes. In the simplest terms, it basically flattens a biological experiment down to one number that answers how much a drug alters a specific cell’s transcriptome.
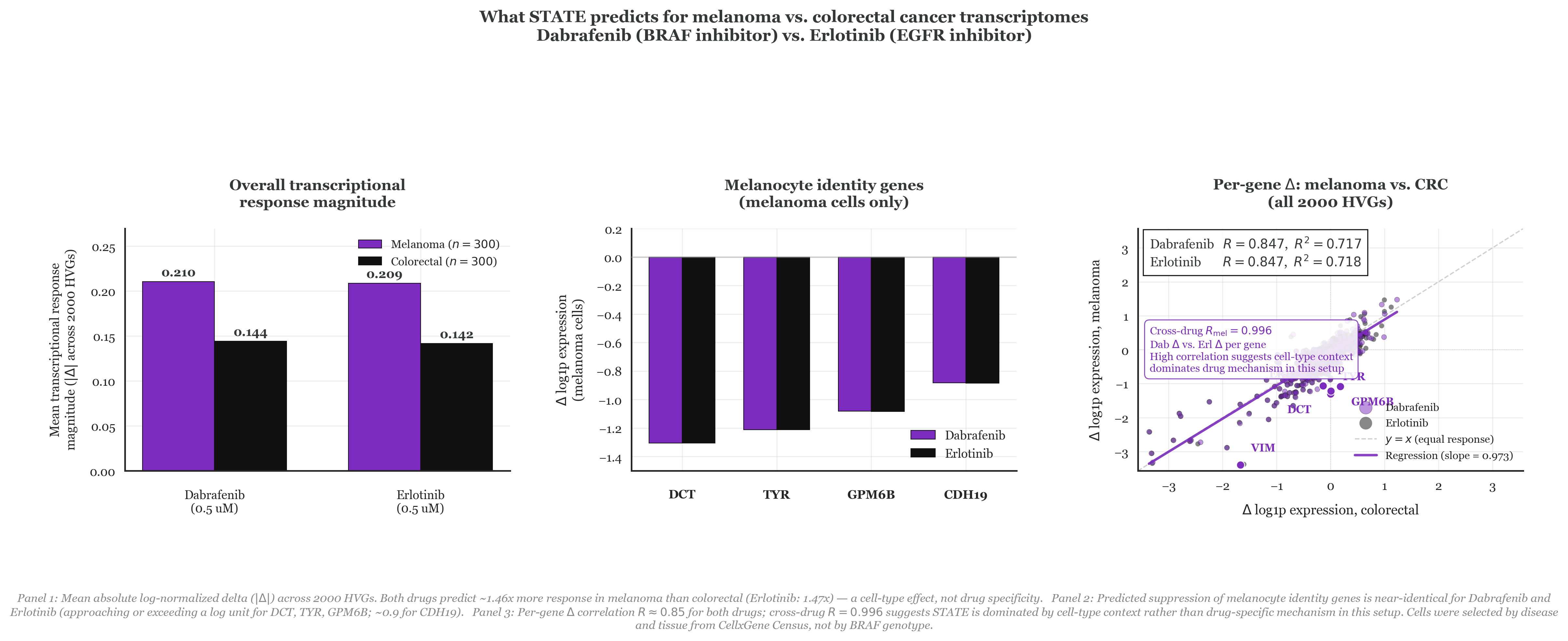
To my surprise, STATE did pick up a difference! Dabrafenib produced 46% more transcriptional disruption in melanoma than in colorectal cells (mean Δ of 0.21 vs. 0.14, as seen in Panel 1). Zooming into melanocyte identity genes specifically (DCT, TYR, GPM6B, CDH19), the model predicts Dabrafenib knocks all of them down by more than a log unit in melanoma (Panel 2). On the surface, that looks like the model correctly reading that Dabrafenib is doing something specific in a BRAF-dependent melanocyte lineage.
But we do REAL SCIENCE here, so I did add a control. I ran the same experiment with Erlotinib, an EGFR inhibitor that washed out in Phase 2 melanoma trials because EGFR isn’t a meaningful driver there. STATE predicted nearly identical results to the clinically useful drug Dabrafenib, showing 47% more disruption in melanoma along with the same suppression of melanocyte identity genes, and a gene-by-gene correlation of R = 0.847 for both drugs across all 2,000 genes (Panel 3). To me, it seems like the model wasn’t predicting drug mechanisms but more so doing some reading of cell-type context. Put differently, based on the way STATE understands the world, melanoma cells just respond louder to perturbations than colorectal cells do. It then follows that the results are what they are. Therein is the problem with cell models – it requires generalizing in a way that language models just aren’t able to do right now.
Is this solvable? I don’t know. Probably depends on whether you think LLMs can achieve artificial superintelligence. In any case, I hope this gave you a sense for what kinds of questions are interesting to answer with cell foundation models, and maybe a glimpse at the frontier.
You can find all data wrangling and code associated with this experiment here.
Christie @GetMentalWealth (via Twitter)
Do you think the future of brain disorders (Parkinson’s to schizophrenia) is curing them? Or getting very good at screening and treating them more effectively?
I am weary of using the word “cure” when talking about any chronic disease, but I think that long-term treatment options could become viable for some degenerative conditions soon. I should also say that I think Parkinson’s and schizophrenia are very different diseases, and the things that will enable a Parkinson’s “cure” will not provide much for people dealing with psychotic conditions.
Where biomedical science has succeeded is mostly in the world of cell engineering, which is very helpful when it comes to replacing lost cells in the body. Parkinson’s happens because some people lose a particular subset of neurons in their brain that leads to the classic rigidity, tremor and less commonly known psychiatric symptoms of the condition. Since the lack of a very specific cell type in a very specific location drives the disease process, it then makes sense that replacing these lost neurons should fix the disease, which actually has happened in some patients. Ashlee spoke with the CEO of a company working on this with some success, so I’m hopeful we can replace lost cells, but the degeneration will still continue. Considering that the average patient is diagnosed somewhere around 65 years old, it’s up to you whether you would consider replacement a cure, if those cells only last 5-10 more years. To me it isn’t, but maybe that’s semantics.
The important point about Parkinson’s is that, while we don’t understand the exact reason the substantia nigra die, we at least know that the cells are disappearing. It’s not everything, but it’s not nothing, either. That brings us to schizophrenia.
Schizophrenia is an unfortunate case where we don’t understand the mechanism enough to have a plausible path towards a cure. It’s a disease with both symptoms that are “added” to a person’s reality, termed positive symptoms in the medical community (hallucinations, delusions, disorganized behavior) and symptoms that take away from someone’s reality, called negative symptoms (flat affect, depression, social isolation). Most antipsychotics work via dopamine receptor blockade, which work fairly well at managing positive symptoms. About 70% of all patients experiencing a first time psychotic episode will get relief from the current crop of drugs. Unfortunately we don’t have great solutions at handling negative symptoms. The end result is that patients might not have hallucinations, but they still struggle to hold down a job or feed themselves due to the underlying cognitive architecture inherent to their disease. I have yet to see anything even directionally close to something that can help us both understand an individual’s neuron architecture, let alone rewrite it reliably.
Gviv @Gviv (via Substack)
What does water/dehydration do to the brain at a cellular level?
I just sat for the second part of my US Medical Licensing Exam and this one was actually considered a “high-yield” (meaning high chance of seeing it on the exam) topic. The first thing to understand is that electrolytes dictate fluid dynamics in the body much more than water itself.
Dehydration triggers a shift where water moves from the intracellular space to the hypertonic extracellular environment (blood vessels). In the brain, this results in acute cellular shrinkage as water exits neurons through specialized aquaporin channels. Because the brain is physically constrained by a rigid skull, this sudden volume loss creates significant mechanical tension on the bridging veins that anchor the brain to the dural membranes. If the shrinkage is rapid or severe enough, these vessels can rupture, resulting in an intracranial hemorrhage. The end result is mechanical stress that manifests as basically anything from lethargy to seizures.
You didn’t ask, but I think it’s interesting to consider the clinical implications of correcting this. To defend its volume during prolonged dehydration, the brain initiates a compensatory synthesis of other chemicals (organic solutes like taurine, glutamine, and inositol). These molecules increase the intracellular osmolality to match the salty environment of the blood, allowing the brain to pull water back into the cells and restore its volume. However, this adaptation creates a dangerous osmotic trap during medical intervention. If free water is replaced too quickly with intravenous fluids, the extracellular fluid becomes hypotonic compared to the solute-heavy interior of the adapted brain cells. Water then rushes into the neurons with enough force to cause massive cerebral edema and potential brain herniation. This necessitates the “high-yield” clinical rule of slow sodium correction, ensuring the brain has sufficient time to shed its protective osmoles and avoid a catastrophic rebound in pressure.
Now you’re ready to sit for Step 2.
Jonathan Whitaker @johnowhitaker (via Twitter)
Did DNA synthesis costs stall? What would it take to make laborious cloning obsolete, and when do you think that might happen?
You have no idea how much I hate molecular cloning. It is so pointlessly tedious, but it used to be way worse. Today we have basically idiot proof enzymes that are so efficient I’ve actually managed to stitch things together that shouldn’t actually work.
Though I’m sure it’s possible to get more efficient than Golden Gate or Gibson Assembly, the real gain I think will come when there’s a CRO you can shoot over a large sequence to and say “make this for me.” What that actually would entail is 1) a really good automated lab and 2) cheap synthesis. In my mind, the question becomes whether cloning is needed in a world where synthesis is extremely cheap.
Cloning emerged as a molecular solution to the problem of wanting to recombine and edit existing strands of DNA you have on hand to make other things. This is useful in situations where you have some “base” you like but want to modify just a portion of it in many different ways. You basically strip away the part you want gone, and add in stuff to replace it. This is necessary because as of right now it is not economically or technically feasible to synthesize massive strands of DNA.
It’s not an interesting answer, but I just don’t see how the tedium of the process can disappear without at least having a substantially better (1) than exists in the US or China, and likely (2). Hopefully someone in the US will solve that first, but if not I’m sure the Chinese CROs are on it.
Nicole @elocinationn (via Twitter)
(paraphrasing) Could you eradicate mosquitoes with EMF?
I was really excited to learn about the EMF remote-controlled mice described in a paper that made the rounds a few weeks ago. I was less excited to learn that it might have been faked. I will defer to my friend Richard Fuisz (and guest of Core Memory) here on the specifics, but it looks like maybe some fraud might be happening.
Let us assume for a moment that it is completely real, and think through what it would take to implement such a thing in mosquitoes. For context I worked on engineering mosquitoes as a member of George Church’s lab at Harvard, and published a couple papers on designing new ways of using CRISPR/Cas9 to suppress mosquito populations. Unfortunately, these approaches largely haven’t scaled. There are many reasons for that, but the biggest one is finding a way of propagating a designed gene – EMF sensitivity, in this case – through a wild population. When that happens we call it a gene drive.
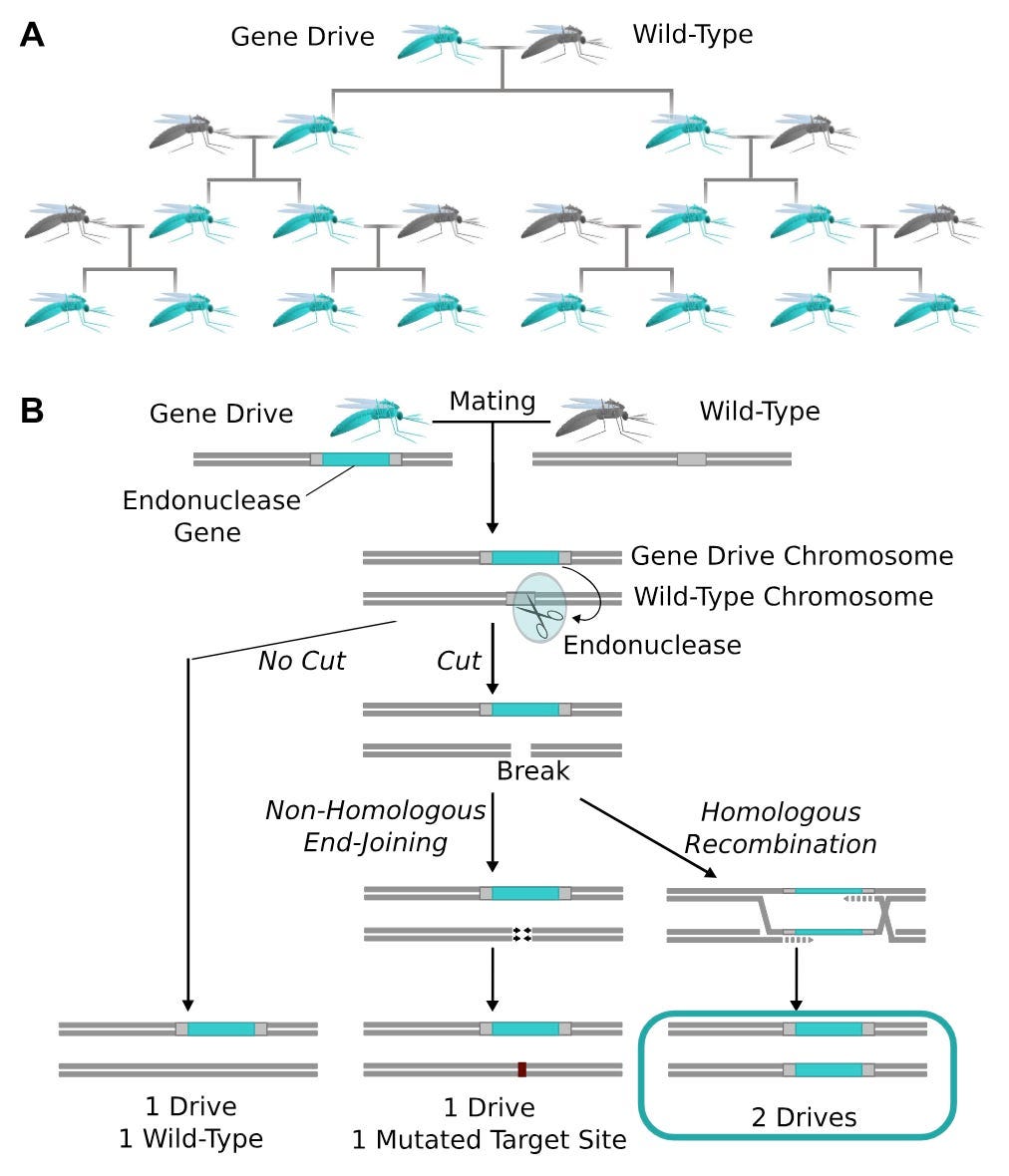
I’m not going to describe the whole thing, as I’ve already done that elsewhere, but the gist is that we can edit mosquito embryos with CRISPR/Cas9 such that the adults that emerge from those embryos have traits we consider desirable for disease control (see figure above for how that works).
The biology hasn’t quite caught up to the engineering plans, though. There are some groups that have successfully demonstrated the ability to drive a gene through a caged population, but it isn’t clear that the gene in the EMF paper has a mosquito-analog. That’s potentially solvable with engineering. Another critical problem is that adult mosquitoes have very particular mating dynamics. They are extremely sensitive to deleterious effects of inserting large genes (which gene drive constructs are) into mosquitoes. Adults come out of this with small wings, thin cuticles, and other stuff, all of which gets them eventually kicked out of the wild population. Hard to build a sustainable gene drive if that’s how your construct is. Also probably resolvable, but much more of a pain than mosquito nets or insecticide coated paint.
To directly answer your question, my guess is this is possible, but likely not going to work as well as we need it to. Buy some DEET and lock in for the summer. Pro tip: if you notice some bites on your body, place a warm rag over the area for a few minutes – it helps kill the swelling.
 | A guest post by
|